
Håller ni med rättarens kommentar?
Halloj!
Jag fick nyligen retur på en inlämningsuppgift och jag skulle vilja fråga vad ni tycker, dels om mitt svar och dels om rättarens kommentar (som ledde till retur). Uppgiften och mitt (handskrivna svar) framgår nedan:


Min rättare skrev:
I svaret till delfråga d har du blandat ihop kinetik med termodynamik. De data som ges i uppgiften är jämviktsdata (Kd). Jämviktskonstanten Kd kan därför inte användas, inte heller förklaras utifrån kinetik. Steriska hinder vid attacken på karbonylkolet förklarar inte storleken på Kd. Sterisk påverkan på reaktionen mellan nukleofil (här vatten) och elektrofil (här karbonylförening) ger en påverkan på hastigheten, inte på jämviktssammansättningen.
Men är inte själva DEFINITIONEN av jämviktskonstanten kvoten av hastighetskonstaterna i båda riktningarna? Om steriska hinder ger en påverkan på hastigheten så påverkar de väl också jämviktssammansättningen? Borde man därför inte kunna resonera kring hastigheterna i vardera riktning och därigenom dra en slutsats om jämviktskonstanten ?
Sedan att jag inte har "resonerat utifrån VSEPR" ordentligt är en annan sak, men det var inte det jag fick kommentar om.

Ja det är relaterat till kvoten mellan dem, men när jämvikten ställt in sig är hastigheterna lika stora. Och visst påverkas hastigheterna av steriskt hinder, dock är den effekten betydande innan jämvikten ställt in sig, t.ex. när vatten tillsätts till karbonylföreningen. När jämvikt råder är hastigheterna i balans, men bildningen av ämnena är förskjuten åt ena hållet.
Men om man har stora steriska hinder i karbonylföreningen (t.ex. aceton), så kommer man väl automatiskt ha för:
Och definitionen för jämviktskonstanten är ju:
Alltså blir större, ju svårare (relativt sönderfallet av hydratet) det är att få karbonylen att hydratiseras. Är det något som inte stämmer i det resonemanget?

I princip har du rätt i att man kan skulle kunna använda att för att resonera kring hur beror på substituenterna. Det är också helt rätt att ökar minskar ju bulkigare och mer elektrondonerande substituenterna är, och exempelvis är mycket större för aceton än för acetaldehyd.
Men vad händer med ? För mig är det inte helt uppenbart hur beror på substituenterna. På något vis hade du behövt argumentera för varför är ungefär samma (eller större) när substituenterna är bulkiga. Har du något argument för detta?
Mer konkret: Hur föreställer du dig att energi-reaktionskoordinatdiagrammet ser ut för hydratisering av aceton respektive acetaldehyd?
Edit: Som naytte påpekar råkade jag blanda ihop "ökar" med "minskar" i första stycket.
Angående din första paragraf så menar jag att det torde vara tvärtom, att borde minska ju bulkigare och mer elektrondonerande substituenterna är.
Angående så tänker jag denna borde öka ju större alkylkaraktär substituenterna har. Dessa donerar in elektrondensitet i kolet och även syrena. Eftersom syrena blir mindre elektrofila borde de intermolekylära bindningarna (vätebindningar) mellan hydratmolekyler men främst vatten och hydrat minska. Då borde väl sönderfallet påskyndas, eftersom hydratet inte kan stabiliseras av lösningsmedlet lika bra?
På samma sätt så tänker jag att borde minska ju större elektrondragande karaktär substituenterna har.

naytte skrev:Min rättare skrev:
I svaret till delfråga d har du blandat ihop kinetik med termodynamik. De data som ges i uppgiften är jämviktsdata (Kd). Jämviktskonstanten Kd kan därför inte användas, inte heller förklaras utifrån kinetik. Steriska hinder vid attacken på karbonylkolet förklarar inte storleken på Kd. Sterisk påverkan på reaktionen mellan nukleofil (här vatten) och elektrofil (här karbonylförening) ger en påverkan på hastigheten, inte på jämviktssammansättningen.
Men är inte själva DEFINITIONEN av jämviktskonstanten kvoten av hastighetskonstaterna i båda riktningarna?
Jag håller med rättarens kommentar.
Nu är jag inte kemist, men från vad jag minns är jämviktskonstanten definierad som quotient av koncentrationer/aktiviteter.
Jag har inte heller studerat detta superingående, men det finns, såvitt jag vet, flera (i princip ekvivalenta) sätt att definiera jämviktskonstanten. Ett är att använda kinetik och då erhåller man , eller så använder man termodynamik och då kör man på aktivitet och fugacitet. I ett idealt system (med ) så är synsätten identiska. I ett verkligt system är det det termodynamiska synsättet som gäller.
Jag håller med rättaren att man kan inte svara på frågan med kinetiska argument. Det som efterfrågas är att förklara tabellens utseende med hjälp av stereriska/geometriska argument. Samtidigt tycker jag inte att det är helt lätt eftersom både geometriska och elektriska egenskaper varierar mellan värdena.
naytte skrev:Angående så tänker jag denna borde öka ju större alkylkaraktär substituenterna har. Dessa donerar in elektrondensitet i kolet och även syrena. Eftersom syrena blir mindre elektrofila borde de intermolekylära bindningarna (vätebindningar) mellan hydratmolekyler men främst vatten och hydrat minska. Då borde väl sönderfallet påskyndas, eftersom hydratet inte kan stabiliseras av lösningsmedlet lika bra?
På samma sätt så tänker jag att borde minska ju större elektrondragande karaktär substituenterna har.
Om vi vill argumentera för att ökar ju bulkigare substituenterna är så borde du resonera om aktiveringsenergin för reaktionen Hydrat -> Karbonyl + Vatten.
(Det är därför jag uppmanade dig att skissa ett energi-reaktionskoordinatdiagram.)
@oneplusone2 och @Pieter Kuiper: Håller ni inte med om att är en rimlig approximation?
oggih skrev:@oneplusone2 och @Pieter Kuiper: Håller ni inte med om att är en rimlig approximation?
k1/k2 är en kvot. Både k1 och k2 skiljer sig åt mellan reaktionerna. Det hjälper inte speciellt mycket att man kan konstatera at k2 bör vara på ett visst sätt i två fall.
oneplusone2 skrev:k1/k2 är en kvot. Både k1 och k2 skiljer sig åt mellan reaktionerna. Det hjälper inte speciellt mycket att man kan konstatera at k2 bör vara på ett visst sätt i två fall.
Detta håller jag helt med dig om; om naytte ska ro iland detta måste svaret adressera hur k2 beror på substituenterna (vilket jag tror kan bli svårt). Anledningen till att jag frågade var att läste ditt förra inlägg som att du menade att det är fundamentalt omöjligt att svara på en fråga om termodynamik med hjälp av kinetiska resonemang.
oggih skrev:naytte skrev:Angående så tänker jag denna borde öka ju större alkylkaraktär substituenterna har. Dessa donerar in elektrondensitet i kolet och även syrena. Eftersom syrena blir mindre elektrofila borde de intermolekylära bindningarna (vätebindningar) mellan hydratmolekyler men främst vatten och hydrat minska. Då borde väl sönderfallet påskyndas, eftersom hydratet inte kan stabiliseras av lösningsmedlet lika bra?
På samma sätt så tänker jag att borde minska ju större elektrondragande karaktär substituenterna har.
Om vi vill argumentera för att ökar ju bulkigare substituenterna är så borde du resonera om aktiveringsenergin för reaktionen Hydrat -> Karbonyl + Vatten.
(Det är därför jag uppmanade dig att skissa ett energi-reaktionskoordinatdiagram.)
Sett till aktiveringsenergi så tänker jag att denna bör minska om substituenterna har alkylkaraktär. Anledningen till detta är att det krävs mindre energi att "bryta" hydrat-vatten-interaktionerna. Om grupper med alkylkaraktär donerar in elektrondensitet blir syrena mindre elektrofila och vätena får då mindre vätejonkaraktär.
Vi vet ur Arrhenius ekvation att om aktiveringsenergin minskar, så ökar hastighetskonstanten.
På motsvarande sätt borde aktiveringsenergin öka om man har elektrondragande substituenter, eftersom detta istället förstärker interaktionerna mellan hydrat- och vattenmolekylerna och gör att det krävs större energi att bryta dessa.
Ur Arrhenius ekvation vet vi att högre aktiveringsenergi leder till en lägre hastighetskonstant.
Angående energidiagramet så borde väl detta huvudsakligen innebära att "toppen" där vi har vårt transition state blir mindre.
Har inte övergångstillståndet (och för den delen även karbonylföreningen) liknande interaktioner med vattnet?
Har inte övergångstillståndet (och för den delen även karbonylföreningen) liknande interaktioner med vattnet?
Jo, men styrkan på vätebindningarna minskar väl i övergångstillståndet? Om jag inte helt ute och cyklar (vilket är möjligt) så ser väl övergångstillståndet ut så här:
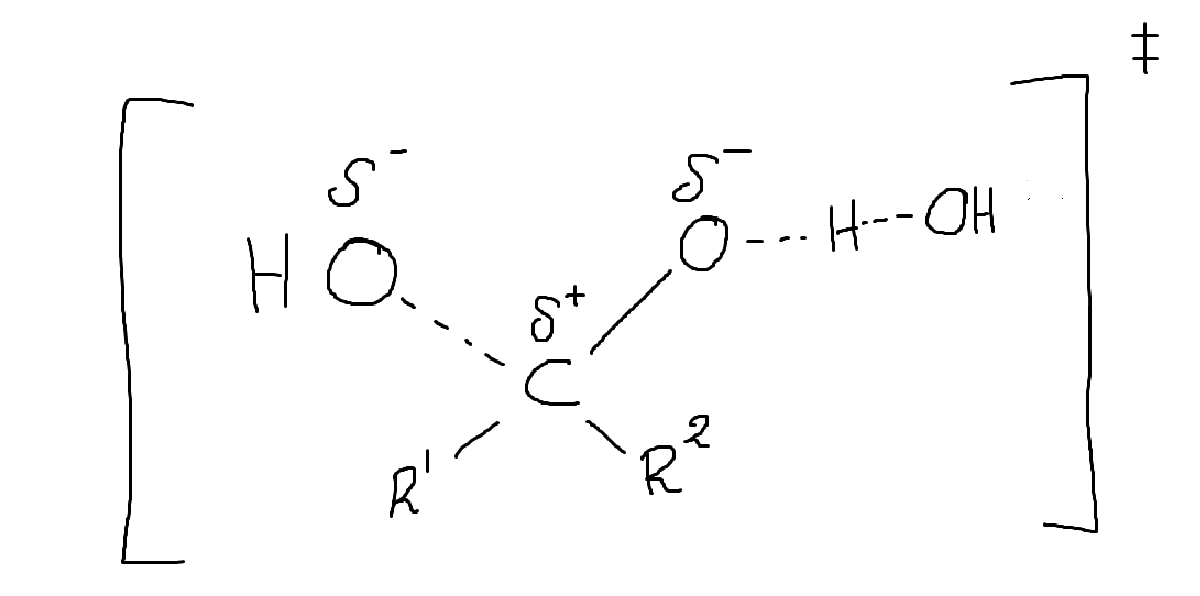
En av hydroxigrupperna har ju i princip stuckit, och ett av vätena håller på att stjälas av en bas. Detta övergångstillstånd kan väl inte rimligtvis ha lika starka interaktioner med vattnet som hydratet?
Angående just karbonylföreningen så borde väl två vätebindningar mellan -OH och vatten i hydratet ge starkare interaktioner än en dipol-dipolinteraktion mellan karbonylsyret och vatten?
naytte skrev:bump
Fråga Smaragdalena, hon kunde kemi.
naytte skrev:Har inte övergångstillståndet (och för den delen även karbonylföreningen) liknande interaktioner med vattnet?
Jo, men styrkan på vätebindningarna minskar väl i övergångstillståndet? Om jag inte helt ute och cyklar (vilket är möjligt) så ser väl övergångstillståndet ut så här:
En av hydroxigrupperna har ju i princip stuckit, och ett av vätena håller på att stjälas av en bas. Detta övergångstillstånd kan väl inte rimligtvis ha lika starka interaktioner med vattnet som hydratet?
Angående just karbonylföreningen så borde väl två vätebindningar mellan -OH och vatten i hydratet ge starkare interaktioner än en dipol-dipolinteraktion mellan karbonylsyret och vatten?
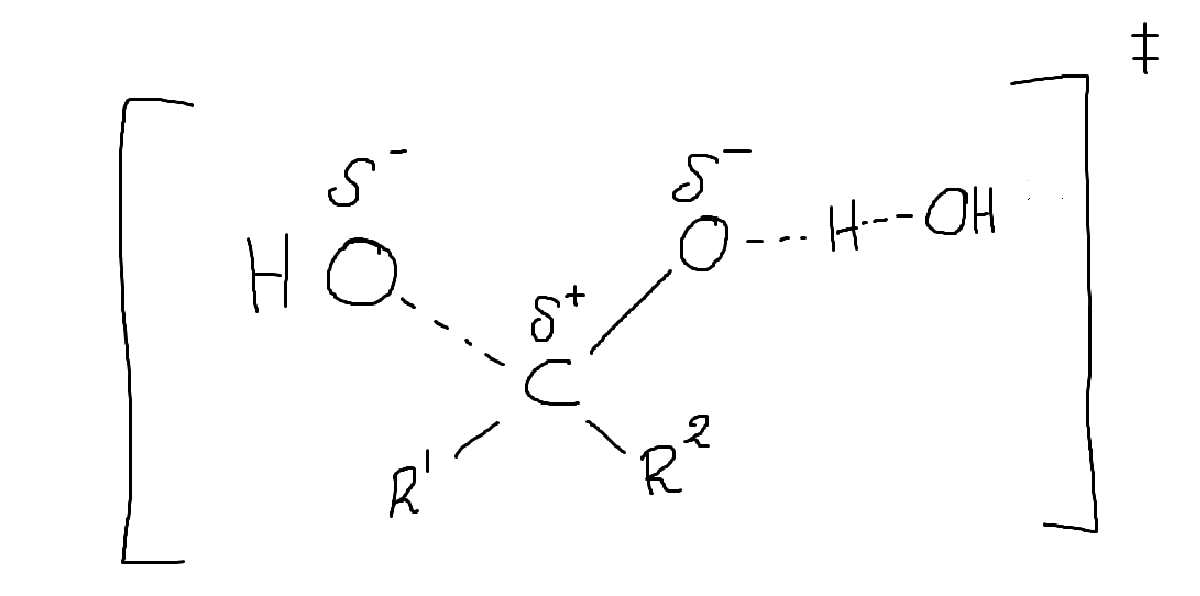
Ditt övergångstillstånd ser inte ut att stämma. Det är ett väte för mycket, och den lämnande (eller attackerande) gruppen kommer vara vatten inte en hydroxigrupp (den är en hyfsat stark nukleofil, och en dålig lämande grupp). Det borde i min mening snarare var liknande nedan (tecknen illustrerar delta +/- inte formella laddningar). Det går också att resonera utifrån att andra vattenmolekyler hjälper till att stabilisera laddningsförskjutningarna.

Angående just karbonylföreningen så borde väl två vätebindningar mellan -OH och vatten i hydratet ge starkare interaktioner än en dipol-dipolinteraktion mellan karbonylsyret och vatten?
Vätebindningar kan skapas från karbonykolet också, inte bara de svagare dipol-dipol bindningarna. Det oladdade karbonylsyret kan agera som vätebindingsacceptor men inte donator. Dock kommer vätebindningen från karbonylsyret att ha en karaktär som är mer eller mindre lik vätebindningarna som uppstår mellan vatten, beroende på vilka substituenterna är d.v.s. om de är elektrondonerande eller -dränerande.
Hej igen och tack för svaren!
Jag har försökt revidera mitt svar enligt min rättares anvisningar. Tycker ni att det ser OK ut nu?

Kanske inte skulle påstå att karbonylkolet i trikloroaetatet destabiliseras. Mer att det blir ännu mer delta-positivt, vilket gynnar attacken mer än i t.ex. fallet med acetaaldehyden, men det misstänker jag var vad du tänkte. Destabilisera associerar mer till ett sönderfall av något som redan finns (som hydraten), snarare än bildandet av något annat.
För att först återkoppla på ursprungsfrågan tycker jag din förklaring låter vettig (även om det är lite spekulativt hur vätebindningarna mellan vattnet och övergångstillståndet fungerar), så jag tycker du har demonstrerat att det går att resonera i termer av hastighetskonstanter.
Men jag tror också tråden har gjort det det tydligt att det är ganska bökigt att behöva förhålla sig till ett teoretiskt övergångstillstånd mellan karbonylföreningen och hydratet, när det egentligen bara är skillnaden i fri energi mellan karbonylföreningen och hydratet som spelar roll 🙂
När det gäller frågan om varför jämviktskonstanten är mindre för kloracetaldehyden jämfört med acetaldehyden så blir jag ärligt talat lite ställd. Jag hänger heller inte riktigt med i era förklaringar.
Ni hänvisar båda till att kloratomen är elektronegativ och gör kolet mer delta-positivt, men exakt hur leder det till att för reaktionen Hydrat -> Karbonyl blir mer positiv?
oggih skrev:När det gäller frågan om varför jämviktskonstanten är mindre för kloracetaldehyden jämfört med acetaldehyden så blir jag ärligt talat lite ställd. Jag hänger heller inte riktigt med i era förklaringar.
Ni hänvisar båda till att kloratomen är elektronegativ och gör kolet mer delta-positivt, men exakt hur leder det till att för reaktionen Hydrat -> Karbonyl blir mer positiv?
Vet inte hur delta G påverkas (om det påverkas alls), men för klorvarianterna kan man väl anta att delta G skulle vara lägre. Vi har endast dissociationskontanten för hydrates sönderfall att utgå ifrån, och saknar information om skillnader i G, entropi, entalpi etcetera, så det blir spekulativt. Mitt resonemang utöver VSEPR för att förklara skillnader i Kd var induktiv effekt. VSEPR kan användas enskilt då klorosubstitenterna är "större", och risken för ogynnsamma interaktioner mellan klors elektroner och de båda hydroxigruppernas i hydratet är högre, så repulsionen borde även den bli högre.
Utöver repulsionen bör induktion även påverka, men jag vet inte hur stor denna effekt skulle kunna vara. Återigen begränsat av brist på de andra parametrarna, men induktionsresonemanget krävs inte heller för att besvara frågan .
Jag tror vi pratar förbi varandra lite här. Målet är ju att förklara K-värdena utifrån strukturen, och eftersom det gäller att K=exp(-DeltaG0/RT) så tänker jag att en tillfredsställande förklaring på något sätt måste adressera hur de strukturella skillnaderna påverkar DeltaG0. Det är väl ändå mindre spekulativt att relatera strukturen till den fria energin, än att i mer lösa termer försöka resonera direkt runt K-värdena?
När det gäller bulkigheten hos kloratomen så tycker jag man kan argumentera för att den har större energihöjande påverkan på hydratet än för karbonylen, eftersom bindningsvinklarna är lägre i hydratet. Alltså kommer kloratomen rent steriskt leda till att DeltaG0 för Hydrat->Karbonyl minskar och att K ökar.
Alltså måste den elektroniska effekten av att kloratomen är elektrondragande på något sätt leda till att DeltaG0 ökar på ett sätt som mer än väl kompenserar för ovanstående.
Jo det verkade vara ett mål. Jag menade inte att det är omöjligt att resonera kring delta G som en tankenöt, men det kräver en del antaganden. Lite av djävulens advokat nu, men delta G behöver ju inte ändras överhuvudtaget, förändringar i entropi kan kompenseras av förändringen hos entalpin.
Red: förtydligade
mag1 skrev:Lite av djävulens advokat nu, men delta G behöver ju inte ändras överhuvudtaget, förändringar i entropi kan kompenseras av förändringen hos entalpin.
Sant! Men nu vet vi ju från datan att är lägre för kloracetaldehyden än för acetaldehyd, så vi vet ju från empirin att för Hydrat->Karbonyl+Vatten ökar vi närvaro av kloratomen, och frågan vi står inför är hur man skulle kunna ha förutsett detta på förhand.
Helt klart har det ju något att göra med det faktumet att klor är elektronegativt, men jag lyckas inte riktigt förstå hur närvaron av en elektrondragande substituent får differensen (Fri energi i karbonylen och vattnet) - (Fri energi i hydratet) att bli mer positiv.
