
Hur fungerar en elektrolytisk cell med en galvanisk cell som spänningskälla?
Halloj!
Jag håller på att studera grundläggande elektrokemi och håller på att titta på galvaniska celler och elektrolytiska celler. Jag vill inleda den här tråden med att beskriva hur jag förstår hur galvaniska celler fungerar, och sedan försöka lirka fram hur det skulle fungera som spänningskälla i en elektrolytisk cell.
Låt säga att vi har den klassiska galvaniska cellen :

Min förståelse för varför elektroner börjar färdas i ledningen har hittills varit: koppar(II)jonerna runt kopparkatoden "snattar" elektroner från denna och reduceras till fast koppar. Då detta snatteri äger rum måste de bestulna atomerna snatta från sina grannar, som i sin tur snattar från sina grannar, ... , tills man har börjat snatta från zinken. Det som gör att man får en mätbar spänning i kretsen är att alla de små elektriska fälten som uppstår till följd av "jonsieringen" som konstant äger rum kan adderas till ett stort elektriskt fält mellan våra två elektroder.
Denna mentala modell har fungerat bra och jag har bekräftat i en tidigare tråd att detta är ett någorlunda korrekt tankesätt.
Min fråga är nu hur denna modell ska tillämpas i en situation som denna:
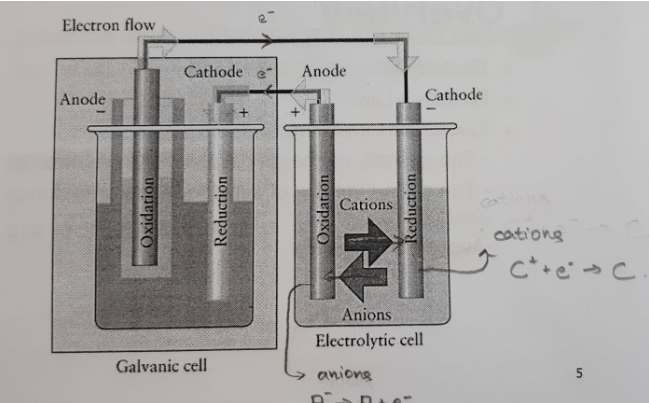
Det som bilden visar är hur en elektrolytisk cell drivs med hjälp av en galvanisk cell, som den ovan. Det jag inte förstår nu är vad som överhuvudtaget får elektronerna att gå i kretsen. Enligt min tankemodell så börjar det breda ut sig ett elektronsnatteri med centrum i den galvaniska cellens katod. Detta snatteri sprider sig till anoden i den elektrolytiska cellen och får där elektroner från ämnet som oxideras. Men sedan förstår jag inte riktigt vad som händer. Vad är det som får katjonerna att reduceras vid den elektrolytiska cellens katod? Det känns som att min modell borde räcka för att beskriva detta, men jag lyckas inte. Här är jag med andra ord lite lost.
Tack på förhand för svar!

Man kan inte riktigt säga att reaktionen börjar i en punkt. Elektronacceptorerna vid ena cellens katod har starkare attraktion (starkare fält) till elektronerna hos elektrondonatorerna vid den andra cellens anod, därav kan elektronerna färdas den vägen. Strömmen kan gå via elektrolyterna då dessa innehåller joner som jämnar ut laddningsskillnader som uppstår vid elektrodreaktionerna. Summerar man de två reaktionerna som sker i respektive cell fås en nettoreaktion med negativ Gibbs energi, dvs elektrontransporten kan ske spontant.
Elektronacceptorerna vid ena cellens katod har starkare attraktion (starkare fält) till elektronerna hos elektrondonatorerna vid den andra cellens anod, därav kan elektronerna färdas den vägen.
Jag förstår inte riktigt den här biten. Skulle du kunna utveckla detta lite eller göra det lite mer konkret kanske genom att vi föreställer oss en cell med bestämda material?
Säg att galvaniska cellen är Ag/Ag+ och Zn/Zn2+ medan elektrolyscellen är Cu/Cu2+ och Ni/Ni2+. Silveratomerna har viss benägenhet att avge elektroner och gå ut i lösning som silverjoner enligt Ag+ + e- <—> Ag. Motsvarande gäller för nickelatomerna i elektroden silverelektroden står förbunden med, fast i ännu högre utsträckning eftersom jämviktsläget för Ni2+ + 2e- <—> Ni ligger längre förskjuten åt vänster. Nickelelektroden, som bevarar elektronerna när nickeljoner går ut i lösning, blir då lite mer negativt laddad än silverelektroden och man får en potentialskillnad, alltså en spänning. Det elektriska fältet som uppstår i ledningen transporterar elektronerna mellan elektroderna. Motsvarande resonemang gäller för Cu/Zn-elektroderna där Zn-elektroden blir negativt laddad. Eftersom det försvinner katjoner vid kopparelektroden och dyker upp katjoner vid nickelelektroden får man en potentialskillnad även i elektrolyten. Ett elektriskt fält uppstår som driver jontransport och leder strömmen genom elektrolyten.
Jag har några frågor om resonemanget du framför här innan jag går vidare: hur "tar sig" elektronerna från kopperelektroden till nickelelektroden? Är det så att elektronerna tvingas på ett species i vattenlösningen som sedan transporterar dessa till anoden?
Eller hur skulle det fungera i just vårt fall? Jag tänker att elektroderna i den elektrolytiska cellen delar en elektrolyt.
EDIT1: eller jag är bara så otroligt förvirrad just nu. Mest på grund av att modellen som fungerat hittills plötsligt inte längre funkar.
EDIT2: eller vänta, jag tror jag fattar nu. Så låt säga att vi vill utföra elektrolys på NaCl. I vår elektrolyt har vi då NaCl (aq), Ni2+ och Cu2+. På grund av potentialskillnaden i ledningen mellan Ag/Ni-elektroderna börjar elektroner sticka till silvret. Då blir det ett överskott av positiv laddning´vid nickelelektroden. Samtidigt tvingar zinket på kopparen elektroner som går ut och reducerar Cu2+ i elektrolyten. Detta skapar ett E-fält även i lösningen, och det är DETTA fält som får Na+ respektive Cl- att röra sig till de respektive elektroderna och reduceras/oxideras?
Ja och vilka joner som huvudsakligen leder strömmen beror på hur mobila de är i lösningen. Joner som binder starkare till vattnet drar med sig ett ”moln” av vattenmolekyler när de rör sig och är därför mindre mobila. I batterier använder man ofta kaliumhydroxid när man vill ha en basisk elektrolyt eftersom kaliumjonen är mer mobil än t.ex. natriumjonen.
Hur stor del av strömmen som transporteras av ett visst jonslag i lösningen brukar kallas för transporttal
Då är jag nog med! Jag blir bara lite fundersam kring vad det innebär att E-fältet "leds" genom ledaren. Det känns lite konstigt att elektroderna kan "känna av varandra" genom ledaren, eller hur man nu ska säga det.
Men nu när jag förstår detta lite bättre så verkar det ju ändå som om idén om elektronsnatteri är tillämpbar här, inte sant? Man får i princip två elektronsnatterier samtidigt, ett snatteri mellan Zn/Cu och ett snatteri mellan Ag/Ni. Potentialskillnaden i elektrolyten som uppstår gör att de species vi vill reducera och oxidera rör sig till respektive elektrod.
Ja så kan du tänka. Angående ledaren kan du tänka att atomerna i en metall kan anses ha en enda stor orbital (ledningsbandet). Om en elektron tillhör en atom längst ut i ena änden så tillhör de på något sätt samtidigt även atomerna i andra änden av ledaren.
Om en elektron tillhör en atom längst ut i ena änden så tillhör de på något sätt samtidigt även atomerna i andra änden av ledaren.
Ja men okej, det är ändå ganska rimligt.
En fråga till: är det så att båda elektroderna i den elektrolytiska cellen måste vara olika i en elektrolys eller kan de vara identiska? Jag minns att jag gjorde en elektrolys någon gång på gymansiet, och då tror jag att båda elektroderna var likadana. Men samtidigt skulle väl den elektrolytiska cellen vi har konstruerat här sluta fungera om vi bytte ut dess elektroder mot typ kolelektroder?
Det fungerar med elektroder av samma material.
Det fungerar faktiskt med elektroder av samma material i den galvaniska cellen också, även om de står i kontakt med två lösningar av samma salt (tänk att du har två zinkelektroder och två zinksulfatlösningar i bilden i första inlägget). Det kräver dock att koncentrationerna i de två lösningarna är olika. En sån typ av galvanisk cell kallas koncentrationscell.
Om vi skulle byta ut elektroderna i den elektrolytiska cellen mot kolelektroder, skulle vi behöva ändra något i elektrolyten då eller skulle vi kunna låta den vara?
I det här fallet skulle det gå bra, det brukar vara viktigare att välja rätt elektroder när det sker gasutveckling. Dels kan gasen reagera med elektroden (syrgasutveckling på en kolelektrod är t.ex. inte bra eftersom det bryter ner den under koldioxidutveckling) och dels blir överpotentialseffekter (som starkt beror på val av elektrod) mer påtagliga vid gasutveckling.
När man framställer koppar industriellt använder man normalt rostfritt stål som elektrod (katod), den lyfts upp efter ett tag och då kan man avlägsna lagret med koppar. Vid framställning av zink använder man normalt aluminiumkatoder.
Okej, då var det ungefär som jag misstänkte!
Men nu tror jag att jag förstår den övergripande principen. Jag skulle bara vilja försöka sammanfatta allt för att säkerställa att jag förstår det rätt rent mekanistiskt.
Låt säga att vi har en uppställning där vi driver en elektrolytisk cell med hjälp av en galvanisk cell. Den galvaniska cellen kan vara Zn (s) | Zn2+ (aq) || Ag+ (aq) | Ag (s), och låt säga att elektrolyten har flytande natriumklorid och två kolelektroder. Då vi kopplar ihop cellerna så kommer jämvikten Zn (s) ⇌ Zn2+ (aq) + 2e- skapa ett elektronöverskott i zinkelektroden. Detta skapar givetvis en laddningsgradient och skapar ett elektronflöde från zinkelektroden till kolkatoden. På liknande sätt kommer jämvikten Ag+ (aq) + e- ⇌ Ag (s) orsaka ett elektronunderskott i silverelektroden. Detta kommer orsaka ett elektronflöde från kolanoden till silverelektroden. Eftersom elektroner flödar till kolkatoden och elektroner flödar bort från kolanoden uppstår det ännu ett elektriskt fält i elektrolyten i den elektrolytiska cellen. Detta driver kloridjonerna till anoden och natriumjonerna till katoden där dessa oxideras respektive reduceras.
Tillägg: 8 feb 2025 01:21
Ursäkta för alla skrivfel! Det känns som jag fick en stroke när jag skrev detta. Har varit vaken väldigt länge så tröttheten smyger sig in i texten.
Ja det verkar rätt uppfattat! Spännande exempel dock, elektrolytiska cellen är en smältelektrolys?
Ja, tydligen haha. Just flytande natriumklorid verkar vara ett vanligt exempel i läroböcker så det var det det fick bli.
Jag vill titta på ett exempel till innan jag grönmarkerar den här tråden, men det gör jag imorgon när jag har sovit lite. Kan inte skriva ordentligt just nu; allt är rätt i mitt huvud men det kommer ut helt fel när jag skriver det. Det exemplet jag vill kolla på är elektrolys för att ta bort rost från olika föremål, ty det verkar som en väldigt användbar och häftig tillämpning av elektrolys och elektrokemi.
Återkommer som sagt imorgon. Tack för all hjälp hittills och god natt! :D
Nu har jag funderat en del och tror att jag har kommit fram till något. Nedan försöker jag beskriva hur en elektrolys av en rostig skiftnyckel går till rent mekanistiskt. Stämmer förklaringen nedan?
Låt säga att vi har en rostig skiftnyckel som vi vill göra rent med hjälp av elektrolys. Som elektrolyt väljer vi natriumkarbonat, och vår anod får vara en kolelektrod. För att åskådliggöra situationen har jag tagit mig friheten att göra en liten skiss:

Innan något ens händer så kommer natriumkarbonatet gå in i ett antal jämvikter, varför vi alltså har följande species i vår lösning: CO32-, HCO3-, OH-, H3O+, Na+, H2O och H2CO3. Då vi slår på spänningskällan kommer elektroner börja flöda ut från batteriet till den rostiga skiftnyckeln. Då detta sker börjar några reaktioner vid katoden ske:
2H2O (l) + 2e- → H2 (g) + 2OH- (aq)
FeO(OH) (s) + 3H3O+ (aq) +3e- → Fe (s) + 6H2O (l)
Den översta reaktionen leder till ett visst mekaniskt arbete, där vätgasen hjälper till att lossa på rosten. Den andra reaktionen "avrostar" skiftnyckeln mer direkt genom reduktion av rosten till metalliskt järn.
Vid anoden oxideras vatten till hydroxidjoner enligt reaktionen:
2H2O (l) → O2 (g) + 4OH- (aq) + 4e-
Om vi lägger ihop halvcellsreaktionerna får vi då en nettoreaktion för den elektrolytiska cellen:
4FeO(OH) (s) + 6H2O (l) + 6H3O+ (aq) → 4Fe (s) + 3O2 (g) + H2 (g) + 12H2O
Katodreaktionerna verkar rimliga, vet inte hur mycket vätgasen hjälper till att mekaniskt ta bort rosten men du kanske har läst om det någonstans? Generellt är ju den reaktionen icke-önskvärd eftersom det stjäl en del av strömmen och gör processen mindre effektiv, man säger att strömeffektiviteten sjunker. Om du beräknar halvcellspotentialerna ser du vilken av reaktionerna som är termodynamiskt mer gynnsam, fast i praktiken lär halvcellspotentialen för sönderdelning av vatten vara lite högre än det du räknar fram p.g.a. överpotentialseffekter då det sker gasbildning.
Anodreaktionen stämmer inte riktigt, den är inte balanserad.
Katodreaktionerna verkar rimliga, vet inte hur mycket vätgasen hjälper till att mekaniskt ta bort rosten men du kanske har läst om det någonstans?
Ja, precis. Jag läste det här för ett tag sedan: https://chemistry.stackexchange.com/questions/124077/eletrolytic-rust-removal
Generellt är ju den reaktionen icke-önskvärd eftersom det stjäl en del av strömmen och gör processen mindre effektiv, man säger att strömeffektiviteten sjunker.
Kan man säga att den reaktionen är beroende av hur mycket rost som finns kvar? Jag tänker att om det finns mycket rost så borde rostreduktionen dominera, medan hydrolysen borde dominera annars (eftersom vattenkoncentrationen är typ konstant och hög).
Anodreaktionen stämmer inte riktigt, den är inte balanserad.
Ojsan, jag råkade skriva hydroxidjoner istället för vätejoner. Menade såklart:
6H2O (l) → 3O2 (g) + 4H3O+ (aq) + 4e-
Angående förändringen i Gibbs fria energi skulle jag gärna vilja försöka räkna på den för att bekräfta att den är positiv. Jag hittar dock inga tabellerade data för reduktionen av rost till fast järn. Eftersom Gibbs fria energi är en tillståndsfunktion borde det väl gå att räkna fram ur kända reaktioner, typ som Hess lag kan användas för att beräkna entalpiförändringar? Eller kan man kanske uppskatta reduktionspotentialen?
Kan man säga att den reaktionen är beroende av hur mycket rost som finns kvar? Jag tänker att om det finns mycket rost så borde rostreduktionen dominera, medan hydrolysen borde dominera annars (eftersom vattenkoncentrationen är typ konstant och hög).
Termodynamiskt ska det inte spela någon roll eftersom aktiviteten hos ett rent fast ämne är 1, oavsett hur mycket av ämnet man har. Om du räknar på elektrodpotentialen ser du det tydligare.
Däremot har det nog en betydelse i alla fall. Rosten bör inte vara elektriskt ledande, ökar därmed resistansen i kretsen och man får ett spänningsfall över rostskiktet. Då behöver man öka spänningen för att reaktionen ska ske i någon större omfattning. I takt med att rosten avlägsnas sjunker resistansen, ger minskat spänningsfall och reaktionen med högre elektrodpotential borde då börja ske i större omfattning.
Angående förändringen i Gibbs fria energi skulle jag gärna vilja försöka räkna på den för att bekräfta att den är positiv. Jag hittar dock inga tabellerade data för reduktionen av rost till fast järn. Eftersom Gibbs fria energi är en tillståndsfunktion borde det väl gå att räkna fram ur kända reaktioner, typ som Hess lag kan användas för att beräkna entalpiförändringar? Eller kan man kanske uppskatta reduktionspotentialen?
Du måste slå upp bildnings-Gibbs för alla ämnen i reaktionen och sedan beräkna skillnaden mellan produkter och reaktanter. Sedan förhåller sig Gibbs energi och elektrodpotential enligt DGo=-nFEo. Det bör gå att hitta bildnings-Gibbs för samtliga ämnen.
Du måste slå upp bildnings-Gibbs för alla ämnen i reaktionen och sedan beräkna skillnaden mellan produkter och reaktanter.
Okej, så med andra ord har vi följande?
Ja exakt, det där är förresten egentligen en variant av Hess lag. Bildnings-Gibbs är ju ändringen i Gibbs energi när ämnet i fråga bildas från grundämnena det består av, så det formeln gör är egentligen att lägga ihop alla dessa reaktioner tills man får nettoreaktionen.
Sedan förhåller sig Gibbs energi och elektrodpotential enligt DGo=-nFEo. Det bör gå att hitta bildnings-Gibbs för samtliga ämnen.
Den här mystiska formeln du använder sen är egentligen samma som arbete = spänning*laddning som man lär sig i gymnasiefysiken.
Man kan visa (finns många härledningar på nätet) att ändringen i Gibbs energi motsvarar maximala icke-tryck-volym-arbetet man kan få ut ur ett system.
F är Faradays konstant, vilket motsvarar (absolutbeloppet av) laddningen hos en mol elektroner.
n är antalet mol elektroner som överförs vid reaktionen.
n*F är med andra ord hur stor laddning som överförs vid reaktionen.
n*F*E blir hur stort arbete som utförs, dvs laddningen multiplicerat med spänningen mellan elektroderna.
Minustecknet är där eftersom arbete utförs på omgivningen, så det försvinner energi från systemet.
Okej, då ger jag det ett försök. Enligt några sökningar är den vanligaste typen av FeOOH i rost α-FeOOH så det är den jag utgår ifrån nu. Enligt sökningar på internet har vi:
Så vi har då:
Här ser vi alltså att , vilket innebär att reaktionen som förväntat inte är spontan.
Jag tror det fortfarande är något fel i balanseringen, väte stämmer väl t.ex. inte?
(Det är för övrigt en av anledningarna till att man normalt förkortar H3O+ till H+, formeln blir annars ganska grötig och man gör lättare fel)
Det har du helt rätt i. Laddningarna stämmer ju inte heller...!
Nu har jag försökt balansera reaktionerna noggrant och kommer fram till:
2FeOOH (s) + 8H2O (l) → 2Fe (s) + 3H2 (g) + 3O2 (g) + 6OH- (aq) + 6H+ (aq)
Det känns även som om man borde kuna summera hydroxiden och väten i HL till vatten, men är lite osäker.
Jag funderade lite hur vätgasen kommer in i bilden, men inser nu att du har lagt ihop bägge katodreaktionerna med anodreaktionen - stämmer det? Det blir inte riktigt rätt, i så fall antar du att de skulle ske i lika stor utsträckning. Du har egentligen två möjliga nettoreaktioner, en för vardera katodreaktion. Sedan kan du beräkna ändringen i Gibbs energi och se vilken som ger störst sänkning, eller annorlunda uttryckt - motsvarar högst potential. Denna reaktion är den som borde ske strikt termodynamiskt.
Tillägg: 8 feb 2025 17:15
Eller ja, eftersom det är elektrolys är det den reaktion med minsta ökning av Gibbs energi som borde ske (minst negativ potential)
men inser nu att du har lagt ihop bägge katodreaktionerna med anodreaktionen - stämmer det?
Japp, det var det jag gjorde. Men jag är nog med på varför det inte fungerar; om den ena delreaktionen bara utgör 0.1 % av halvcellsreaktionerna som äger rum vid katoden så blir den nettoreaktionen jag har tagit fram inte representativ för vad som händer.
Nu har jag försökt ta fram de möjliga nettoreaktionerna:
(i) 4FeOOH (s) → 4Fe (s) + 3O2 (g) + 2H2O (l)
(ii) 6H2O (l) → 2H2 (g) + O2 (g) + 4H+ (aq) + 4OH- (aq)
Ser dessa rätt ut?
Tillägg: 8 feb 2025 19:16
Ursäktar dessutom för att mycket av det jag skriver är lite förvirrat. Har extremt mycket otur när jag tänker idag (lågt blodsocker). Ska åtgärda problemet nu genom att trycka i mig bröd.
(i) är rätt
(ii) är också rätt men om du lägger ihop H+ och OH- till H2O kan du förkorta bort ett gäng H2O i formeln. Det är okej att lägga ihop dem om man har en gemensam elektrolyt, men om man har två separerade lösningar så får man en sur respektive basisk lösning.
Så (ii) kan med andra ord skrivas som 2H2O (l) → 2H2 (g) + O2 (g)?
Jo det stämmer
Okej, då gör jag ännu ett försök med Gibbs fria energi:
Om vi nu beräknar reaktionernas cellpotentialer har vi:
Så eftersom (i) har en större potential så är den mer termodynamiskt gynnsam och kommer därför äga rum i större utsträckning? Mina antal elektroner tog jag från mina balanserade halvcellsreaktioner (den balanseringen som krävdes för att jag skulle kunna lägga ihop halvcellsreaktionerna).
(i) kommer termodynamiskt vara den enda reaktionen som sker, men i praktiken observerar du bägge reaktionerna där (i) dominerar.
Några kommentarer:
- Enheten hos delta G blir kJ, inte kJ/mol (2 och 4 ska egentligen vara 2 mol och 4 mol). Du ser också att det blir fel enhet för potentialen, där du nu får enheten J/(mol*C) istället för J/C (den sistnämnda motsvarar enheten V).
- Jag tror du har räknat med fel n för (ii), visst är det fyra och inte tre elektroner som överförs?
Enheten hos delta G blir kJ, inte kJ mol (2 och 4 ska egentligen vara 2 mol och 4 mol). Du ser också att det blir fel enhet för potentialen, där du nu får enheten J/(mol*C) istället för J/C (den sistnämnda motsvarar enheten V).
Yes, märkte det precis också!
Jag tror du har räknat med fel n för (ii), visst är det fyra och inte tre elektroner som överförs?
Stämmer bra. Då blir det dock lite konstigare, för om man räknar med rätt antal överförda elektroner får man ungefär samma potential som för (i). Betyder det att reaktionerna kommer ske i ungefär samma utsträckning? Jag får:
(missade att skriva till prefixet kilo i #31)
Ja då blir de ungefär lika termodynamiskt (o)gynnsamma. Överpotentialen kommer avgöra vad som sker i praktiken, troligen kommer (i) fortfarande dominera eftersom du där har gasutveckling på en elektrod medan det sker på bägge elektroderna i (ii). Många fler faktorer styr också värdet på överpotentialerna, t.ex. storleken på diffusionslager intill elektroderna.
Sen ska man komma ihåg att det du har beräknat är potentialerna vid normaltillstånd (normalpotentialer), dvs 1 bar, 25 grader och 1 M av alla ämnen (som motsägelsefullt nog också är oändligt utspädda). Vid andra förutsättningar får man andra potentialer, vilka du kan beräkna med Nernsts ekvation.
Sen ska man komma ihåg att det du har beräknat är potentialerna vid normaltillstånd (normalpotentialer), dvs 1 bar, 25 grader och 1 M av alla ämnen (som motsägelsefullt nog också är oändligt utspädda).
Yes, jag visste att normaltillstånd innebar 1 bar, 25 °C och 1 M av ämnena, men hur kan de vara "oändligt utspädda" om koncentrationen är 1 M? Borde den inte vara infinitesimal i så fall? Eller missförstår jag begreppet "oändligt utspädd"?
Och är inte detta extremt stora normalpotentialer? Behöver man inte ett batteri som kan alstra en så stor spänning för att elektrolysen ska kunna äga rum?
Jo just därför är det motsägelsefullt, det är alltså ett teoretiskt tillstånd som inte kan existera i verkligheten utan man måste via Nernsts ekvation räkna om potentialen för gällande villkor. Jag antar att man har det antagandet för att det då gäller ideal lösning och man kan sätta aktivitetskoeffiecienten till 1.
Det är ganska höga normalpotentialer men det är inget man höjer på ögonbrynen över. Ett vanligt alkaliskt batteri ger väl en spänning på runt 1,5 V och ett litiumjonbatteri över 3 V.
Hehe, ser nu att du har skrivit kV och inte V. Du har missat någon tiopotens i beräkningen, det ska vara V.
När du skriver 96.485, är punkten tusentalsavskiljare eller decimaltecken? F=96485 C/mol.
Ack, jag föll offer för en av jänkarnas konstigheter, att de skriver ut tusentalsavskiljare...
Då blir det inte kV utan bara V, vilket är betydligt mer logiskt!
Men när man säger att alla ämnen ska vara 1 M, avses då alla ämnen lösta i vatten? För vi kan väl inte prata om 1 M rost?
Nej precis, det gäller för lösta ämnen.
Vad bra!
Ändå lite förvånande ändå att det räcker med typ ett litet litiumbatteri för att driva en sådan här process. När jag kollar på nätet så rekommenderar många att man använder ett bilbatteri som kan leverera en spänning på närmare 12 V.
Har du någon aning om varför det rekommenderas så höga spänningar om de som krävs är ungefär en tiondel av dem?
Det är fortfarande bara det termodynamiska behovet för att driva reaktionen som du har beräknat, överpotentialseffekter gör att du behöver en högre spänning i praktiken. Du kan tänka att du har en resistor seriekopplad med spänningskällan och elektrolyscellen.
Ja okej, det är sant. Vattnet och andra material i kretsen höjer ju resistansen.
En liten fråga till jag kom att tänka på i duschen idag:
Om vi skulle föra ned en bit fast zink i en vattenlösning av ZnSO4, då skulle ju zinket ställa in sig i någon slags jämvikt enligt Zn (s) ⇌ Zn2+ + 2e-, inte sant?
Ja så är det
Kan vi rent generellt säga att den jämvikten kommer gynna bildandet av Zn2+-joner under normala förhållanden (1 M, 25 grader osv...)?
Jag vet inte riktigt vad du menar med "gynna", men reaktionen begränsas av att zinkjonerna som går ut i lösning förhindrar ytterligare zinkjoner från att gå ut i lösningen (de repellerar varandra), samtidigt som elektronerna som är kvar i elektroden förhindrar att ytterligare elektroner lämnas kvar. Först när kretsen sluts kan det ske en större transport av elektroner och joner.
Med gynnas menade jag att jämvikten är förskjuten åt höger.
Jag klurade lite kring galvaniska celler efter att ha sovit på det vi diskuterade igår och jag får uppfattningen att det som får strömmen i t.ex. Daniellcellen att gå är att zinken är mer benägen att gå ut i lösning än att reduceras tillbaka till fast zink medan koppar är mer benägen att reduceras till fast koppar än att gå ut i lösning. Det är alltså detta som skapar vår potentialskillnad och alltså vår spänning.
Ja precis, zinkatomerna tenderar att gå ut i lösning och kvarlämna elektroner medan kopparjonerna i större omfattning tenderar ta upp elektroner och lämna ett underskott i andra elektroden.
Hallelujah!
Tack så hemskt mycket för all din hjälp i den här tråden! Tack vare dig har jag lyckats räta ut något jag har haft svårt att förstå ganska länge!!
Riktig king!
